Penulis: Witri Winanda, S.Si. M.Si.
Setiap anak terlahir unik yang membuat setiap manusia berbeda satu sama lain. Ada yang tumbuh cepat, ada pula yang berkembang dengan ritme berbeda karena kondisi genetik tertentu. Beberapa anak terlahir dengan kondisi istimewa salah satunya Seckel Syndrome yang membuat tubuh mereka tampak mungil layaknya boneka. Namun di balik penampilan itu tersimpan kisah genetik yang menakjubkan.Seckel Syndrome kerap disertai mikrosefalus ukuran kepala yang lebih kecil dari normal. Kisah tentang Seckel Syndrome bukan hanya soal kelainan fisik, tapi juga tentang bagaimana sains membantu kita memahami keajaiban perkembangan manusia sejak di dalam rahim.
Apa itu Seckel Syndrom
Seckel Syndrome merupakan kelainan genetik langka dengan angka kejadian sekitar 1 dari 10.000 kelahiran tanpa perbedaan jenis kelamin.yang diturunkan secara autosom resesif. Kondisi ini terekspresi ketika seseorang mewarisi gen bermutasi dari kedua orang tuanya. Sindrom ini menyebabkan gangguan pada pembentukan tulang (osteodisplasia), disertai dengan mikrosefalus, serta postur tubuh kerdil proporsional. Kondisi ini pertama kali diamati pada tahun 1959 oleh ilmuwan Jerman Rudolf Ludwig Karl Virchow. Kemudian di tahun 1960 dijelaskan secara ilmiah oleh dokter anak Helmut Paul George Seckel, yang akhirnya membuat sindrom ini dikenal dengan namanya. Bayi yang lahir dengan kondisi ini biasanya berukuran sangat kecil, memiliki proporsi tubuh seimbang, dan menunjukkan ciri wajah khas seperti hidung runcing, dagu kecil, dan mata besar. Kondisi ini termasuk dalam kelompok primordial dwarfism, yakni gangguan pertumbuhan yang sudah muncul sejak tahap embrio.
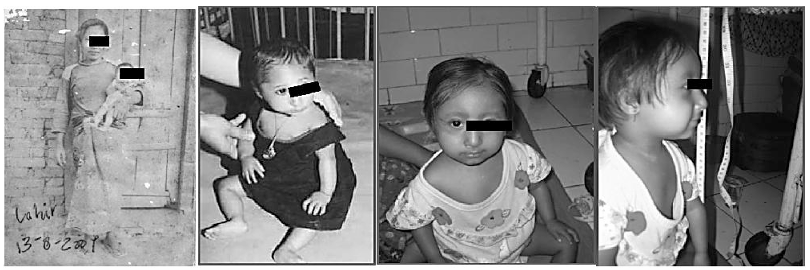
Gambar 1. Ciri Fisik Seckel Syndrome kasus di daerah Lombok (Malino et.al. 2003)
Salah satu tanda yang paling umum pada penderita Seckel Syndrome adalah mikrosefalus kondisi ketika ukuran kepala jauh lebih kecil dari ukuran normal anak seusianya. Mikrosefalus terjadi karena otak tidak berkembang sempurna selama masa kehamilan. Ini terjadi karena gangguan pembelahan dan diferensiasi sel otak selama perkembangan janin. Kondisi ini bisa disebabkan oleh banyak faktor; mulai dari infeksi selama kehamilan, paparan zat berbahaya, hingga mutasi genetik seperti pada Seckel Syndrome. Seckel Syndrome diwariskan secara autosomal resesif, yang berarti seorang anak hanya akan mengalami sindrom ini jika menerima salinan gen bermutasi dari kedua orang tuanya. Orang tua yang membawa satu salinan gen tersebut tidak menunjukkan gejala, tetapi berpotensi menurunkannya pada keturunan mereka.
Dalam sebagian besar kasus, diagnosis Seckel Syndrome didasarkan pada pengamatan klinis terhadap tanda dan gejala khas. Pada beberapa kasus, pernah dilaporkan adanya peningkatan jumlah kerusakan kromosom (chromosomal breakage), namun temuan ini tidak selalu muncul pada semua penderita, sehingga tidak dapat dijadikan acuan utama dalam penegakan diagnosis. Ciri-ciri yang tampak pada pemeriksaan X-ray (rontgen) meliputi usia tulang yang tertunda (retarded bone age), serta kelainan bentuk panggul (displasia panggul) dan pergeseran kepala tulang radius (dislokasi kepala radius). Pada Kasus balita perempuan di daerah Lombok menunjukkan hasil analisis kariotipe kromosom 46XX, yang menandakan pasien berjenis kelamin perempuan tanpa adanya kelainan struktural kromosom utama. Temuan ini mengindikasikan bahwa gangguan yang dialami bukan disebabkan oleh kelainan kromosom besar seperti delesi atau translokasi, melainkan kemungkinan berasal dari mutasi genetik spesifik yang lebih kecil. Pada kasus Seckel Syndrome ini, mutasi umumnya terjadi pada gen ATR, CENPJ, dan CEP152 yang berperan penting dalam mekanisme perbaikan DNA serta pengaturan siklus sel.
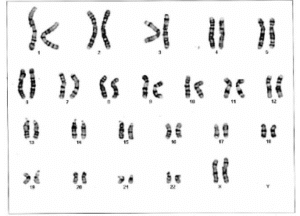
Gambar 2. Analisis kromosom menujukkan tidak ada kelainan struktural utama (Malino et.al. 2003)
Hingga kini, para peneliti telah mengidentifikasi beberapa gen penting yang berkaitan dengan Seckel Syndrome. Sindrom ini diketahui diturunkan secara autosomal resesif akibat ketidakstabilan kromosom. Beberapa lokus genetik (SCKL1–SCKL5) telah dipetakan, dan tiga gen utama yang berperan telah diidentifikasi, yaitu ATR (Ataxia Telangiectasia-Related Protein), CENPJ (Centromere Protein J), dan CEP152 (Centrosomal Protein 152 kDa). Ketiga gen tersebut berfungsi dalam respon seluler terhadap kerusakan DNA dan regulasi pembelahan sel. Mutasi pada gen-gen ini menyebabkan gangguan dalam proses pembelahan sel, terutama di jaringan otak dan tulang. Akibatnya, pertumbuhan tubuh dan otak terhambat sejak tahap embrio. Seckel Syndrome termasuk kondisi yang sangat bervariasi secara genetik (heterogen). Karena sifatnya yang sangat beragam, para peneliti memperkirakan mutasi gen lain yang berhubungan dengan sindrom ini mungkin akan ditemukan di masa depan.
Editor: Ambarwulan, S.T.
Referensi
- Chentli, F.C., Belahcene, S., & Azzoug S. 2013. Seckel’s -like Syndrome with primordial dwarfism, marke retardation and severe heart malformation. Ibnosina J.Med. BS. 339-344.
- Hall, J. G., Flora, C., Scott, C. I., Pauli, R. M., & Tanaka, K. I. 2004. Majewski osteodysplastic primordial dwarfism type II (MOPD II): Natural history and clinical findings. American Journal of Medical Genetics Part A, 130A(1), 55–72.
- Malino, I., Y., Arimbawa, M., & Surya, B. 2003. Seckel Syndrome in a-2 year old girl. Jurnal Imiah Kedokteran. 62-67.
- O’Driscoll, M., Ruiz-Perez, V. L., Woods, C. G., Jeggo, P. A., & Goodship, J. A. 2003. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nature Genetics, 33(4), 497–501.
- Qvist, P., Huertas, P., Jimeno, S., Nyegaard, M., Hassan, M. J., Jackson, S. P., & Westphal, D. 2011. The genome maintenance protein SPIDR links the Bloom syndrome helicase and homologous recombination factors. Nature Cell Biology, 13(6), 623–629.
- von der Hagen, M., Pivarcsi, M., Liebe, J., von Bernuth, H., Didonato, N., Hennermann, J. B., & Kaindl, A. M. 2014. Diagnostic approach to microcephaly in childhood: A two-center study and review of the literature. Developmental Medicine & Child Neurology, 56(8), 732–741.